

科研进展
兰州化物所分子识别催化材料研究获进展
高活性、高选择性的多相催化材料创制是催化研究领域的重要目标之一。在众多选择性调控手段中,基于均多相融合理念构筑金属-有机活性表界面是提高催化反应选择性的有效技术。以往的研究中,催化剂选择性的提高多数以牺牲表面活性位点数量或催化活性为代价。通过分子印迹策略设计制备一类能同时提高催化剂活性和选择性的均多相融合催化材料是一项值得研究的课题。
中国科学院兰州化学物理研究所羰基合成与选择氧化国家重点实验室均多相融合催化课题组近年来致力于表面配体构筑研究(Nat. Commun. 2019, 10, 2599-2605; J. Catal. 2021, 400, 397-406;分子催化, 2023, 37, 213-224),重点关注配体与活性金属的相互作用、表面新活性位点的构建。
近日,该课题组通过在Cu/Al2O3表面上依次吸附印迹分子(N)和印迹配体(L)的方法制备了一系列分子印迹催化剂(Cu/Al2O3-N-L,图1,a)。该类催化材料对硝基化合物中的印迹分子表现出较高的加氢活性和独特的分子印迹效应(图1,b)。通过结构表征和DFT计算,研究人员发现该催化体系具有优异分子印迹效果,其原因是形成了新的活性Cu-N位点和精确的分子印迹空腔。
该催化体系具有活性位点结构明确、易于循环利用、制备方法具有一定普适性等特点,为合成具有新活性位点和精确印迹腔结构的分子印迹催化剂提供了一种新的思路。

图1. Cu/Al2O3-N1-L1分子印迹催化剂的制备过程(a)、催化剂活性结构及其与Cu/Al2O3活性对比(b)示意图
一系列催化剂结构表征表明,分子印迹催化剂表面的金属Cu与印迹配体L1之间存在电子相互作用,形成了稳定的Cu-N位点(图2,a-g),同时差分电荷密度图也证实了Cu-N之间的电子转移(图2,h)。
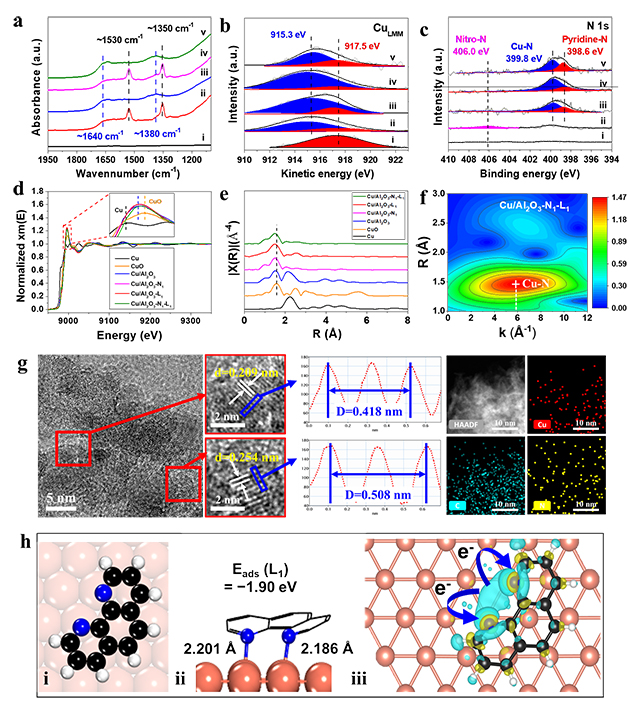
图2. Cu/Al2O3-N1-L1催化剂结构表征结果(a-g)和Cu-N位点的差分电荷密度图(h)
进一步DFT计算发现,在印迹分子和印迹配体依次吸附在Cu表面后,Cu与N之间的相互作用构筑的Cu-N活性位点和分子印迹空腔是硝基化合物加氢活性和选择性提高的根本原因。印迹空腔在对分子存在择形效应的同时还有利于印迹分子的优先吸附(图3,a-h),Cu-N位点有利于H2的活化解离,两者协同作用提高了催化剂的活性和选择性(图3,i-j)。

图3. 多种硝基化合物在不同印迹空腔中的吸附能对比(a-h)和H2在不同位置的解离能对比(i-j)
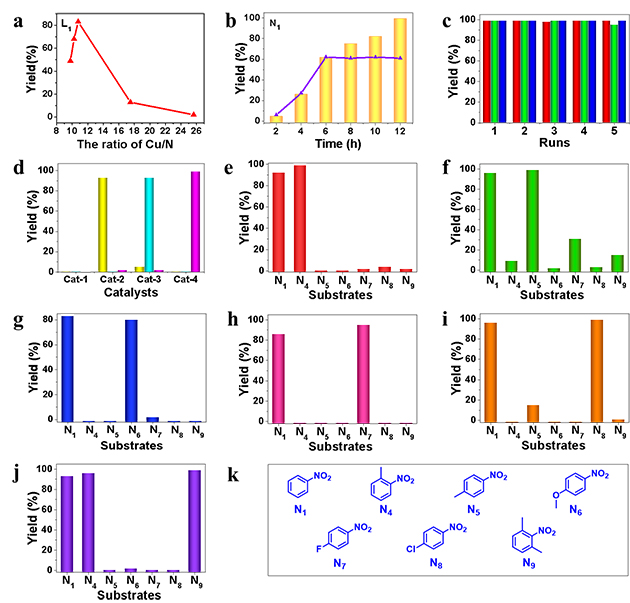
图4. 催化剂的时间曲线和重复使用(a-d)和不同印迹分子构筑的分子印迹催化剂的催化性能(e-k)
研究人员还发现,该分子印迹催化剂可重复使用多次且相应的分子印迹功能得到保留(图4,c-d)。此外,通过更换印迹分子可制备出多种用于其他硝基化合物高选择性加氢的分子印迹催化剂,表明该方法具有较好的普适性(图4,e-k)。
上述研究工作以“Construction of Highly Active and Selective Molecular Imprinting Catalyst for Hydrogenation”为题发表在J. Am. Chem. Soc.(DOI: doi.org/10.1021/jacs.3c04576)上,何东城博士为该论文第一作者,石峰研究员和崔新江研究员为共同通讯作者。
以上工作得到了国家自然科学基金、甘肃省自然科学基金和甘肃省重大项目等的支持。





